{kind=link}
Distrofi muskulore Duchenne (mdd)
Dystrofia muskulare e Duchenit është një nga format më të zakonshme të kësaj sëmundjeje. Sëmundja bëhet e dukshme rreth vitit të dytë të jetës dhe ndodh vetëm tek djemtë, e cila shoqërohet me llojin trashëgimi të recesionit të lidhur me X. Simptomat e mëposhtme janë tipike për DMD.
■ Dobësi muskulore. Bëhet e dukshme kur fëmija ka vështirësi në lëvizje ose lëvizje gjymtyrësh. Fëmija mund të fillojë të ecë në këmbë, nuk mund të ngjitet në shkallë, të ngrihet në këmbë vetëm me ndihmën e duarve. Simptoma e fundit që vjen nga dobësia e muskujve të dyshemesë së legenit quhet simptomë Gauer.
■ Megjithëse muskujt nuk vuajnë nga pushimi dhe nuk ka dhimbje kur shtypen, pacienti bëhet i vështirë për të kryer veprime të caktuara. Muskujt e prekur janë të dobët, por shpesh shfaqen të zgjeruara - ky fenomen quhet pseudohypertrophy.
■ Kufizimi i mobilitetit. Karakteristikë për fazat e fundit të DMD. Shpesh ndodh që, kur disa muskuj të dobësohen, muskujt e tyre antagonistë mbeten të fortë dhe fëmijët e sëmurë fillojnë, për shembull, të ecin në majë të këmbës. Është e vështirë për të ruajtur pozicionin e trupit dhe pacientët mund të kërkojnë një karrocë.
■ Pacienti zhvillon deformime progresive dhe lakimi i kockave, lodhja dhe deri në moshën 10 vjeç, shumica e pacientëve bëhet me aftësi të kufizuara. Pacientët zakonisht vdesin para moshës 20 vjeçare. Shkaku i vdekjes është një infeksion pulmonar, i shoqëruar me dobësi të muskujve të frymëmarrjes ose me arrestimin e zemrës.
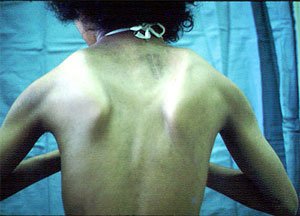
Format e pazakonta të distrofisë muskulare
Ka një numër të llojeve të tjera të distrofisë muskulare. Dystrofia muskulore e Becker është një sëmundje e lidhur me kromozomin X, më dashamirës se Duchenne, që shfaqet në moshën 5 deri në 25 vjet. Njerëzit me këtë lloj distrofi jetojnë më gjatë se me DMD. Dystrofia e brezit të shpatullave ndodh me të njëjtën frekuencë në personat e të dy gjinive dhe zakonisht manifestohet në moshën 20-30 vjeçare. Përafërsisht 50% e njerëzve që vuajnë nga ky lloj i distrofit, dobësia shfaqet në brezin e humeralit dhe nuk mund të përhapet në rripin e skajit të skajit, ndërsa në të tjerat muskujt e barkut të krahut të poshtëm janë prekur së pari dhe dobësia në brezin e shpatullave shfaqet pas rreth 10 vjetësh. Kursi i sëmundjes është zakonisht më i mirë në ato pacientë që fillimisht kanë gjymtyrë të sipërme. Dystrofia muskulare e fytyrës së krahut është e trashëguar nga një mekanizëm dominant autosomal dhe ndikon në mënyrë të barabartë fytyrat e të dy gjinive. Mund të ndodhë në çdo moshë, por zakonisht shfaqet për herë të parë në adoleshentë. Ky lloj i distrofisë karakterizohet nga skarpula "pterygoid". Disa njerëz kanë një lordozë të fortë mesit (lakimi i shtyllës kurrizore). Dobësia e muskujve të fytyrës çon në faktin që njerëzit nuk mund të fishkëllejnë, të tërheqin buzët e tyre ose të mbyllin sytë. Varësisht nga grupet e muskujve të prekur, kapja dhe levizjet e vogla të gishtave mund të dobësohen ose mund të shfaqet një "ndalesë e varur". Nuk ka trajtim mjekësor për distrofi muskulare, por ndërlikimet, siç janë infeksionet e rrugëve të frymëmarrjes dhe urinës, kërkojnë antibiotikë.
Trajtimi përfshin aktivitetet e mëposhtme:
■ Ushtrim fizik - kjo mund të ngadalësojë zhvillimin e dobësisë dhe kufizimin e lëvizjes; komplekset e ushtrimit nën mbikëqyrjen e një fizioterapisti janë shumë të dobishme.
■ Shtrirja pasive e tendons, të cilat kanë tendencë të jenë të shkurtuar.
■ Me shfaqjen e deformimeve dhe laktave të shtyllës kurrizore, nevojiten korse korrigjuese.
■ Traction kirurgjik i tendons shkurtuar.
■ Ndihma psikologjike është shumë e rëndësishme; mbështetje jetike për familjen dhe rehati në shtëpi.
Prognoza dhe sëmundshmëria
Në disa raste, sidomos me distrofinë e Duchennes, prognoza e sëmundjes është e pafavorshme. Shkalla e aftësisë së kufizuar mund të jetë shumë e rëndësishme, me kohë pacientët mund të ndalojnë në këmbë. Shumica e pacientëve me distrofinë e brezit të shpatullave mund të ndihmohen për të udhëhequr një jetë të plotë, ndonëse pak të ndryshuar brenda 20-40 vjetëve dhe ndonjëherë edhe më shumë. Njerëzit të cilët zhvillojnë distrofinë muskulore në adoleshencë vonë zakonisht kanë një prognozë më të mirë. Profilaksina e distrofisë muskulare ende nuk është e mundur, megjithëse zbulimi i një gjeneti të dëmtuar ka rritur gjasat e terapisë së gjeneve.
Prevalenca e sëmundjes
Distrofi muskulare është një sëmundje mjaft e rrallë, por është e zakonshme në mbarë botën midis njerëzve të të gjitha racave. Forma më e zakonshme - distrofi muskulare Duchenne - ndodh me një frekuencë prej rreth 3 rastesh për 10,000 djem.
arsyet
Të gjitha llojet e distrofisë muskulare janë shkaktuar nga shkaqe gjenetike, edhe pse shkaku i saktë i degjenerimit të indit të muskujve është i panjohur. Ndoshta shkaku kryesor është një shkelje në membranën qelizore, e cila në mënyrë të pakontrolluar kalon jonet e kalciumit në qelizë, e cila aktivizon proteazën (enzimat) që kontribuojnë në shkatërrimin e fibrave të muskujve. Diagnoza e mundshme prenatale në formën e studimit të lëngut amniotik para lindjes. Megjithatë, prindërit që vuajnë nga distrofia muskulore, para se të kenë një fëmijë, kanë nevojë për këshillim gjenetik mjekësor.
Diagnostics
Rastet tipike të përparimit të ngadaltë janë klinikisht të dukshme. Në pacientët, sidomos me distrofinë e Duchenne, ekziston një nivel i lartë i kreatinës kinazës në gjak. Për të dalluar distrofi nga çrregullime të tjera, mund të jetë e nevojshme të kryhet elektromiografia. Diagnoza zakonisht konfirmon një biopsi; studimet histokimike ndihmojnë në dallimin e distrofisë nga llojet e tjera të miopateve.